Support and Services
Columns Troubleshooting
Reversed-phase liquid chromatography frequently employs organic solvents such as methanol, acetonitrile or tetrahydrofuran. Although HPLC grade products of these solvents are available, it seems some users have trouble when using a reagent grade solvent instead of HPLC grade. This leads them to waste considerable amounts of time. How do the two solvent grades differ?
Methanol/acetonitrile
Reagent grade solvents contain larger quantities of UV absorbing impurities than HPLC grade solvents. This makes it difficult to use reagent grade solvents for gradient elution or trace analysis due to these impurities being retained and eluted as peaks under gradient conditions. This is particularly so when detection is conducted at a short wavelength when significant differences appear in baseline noise or detection sensitivity. In some cases (or at certain specific wavelengths) it may be possible to use a reagent grade solvent but we recommend HPLC grade solvents in order to obtain a stable, reproducible chromatogram.
Tetrahydrofuran
Tetrahydrofuran easily generates peroxides. In order to prevent this antioxidants are frequently added. The antioxidants can cause a ghost peak so a solvent containing no antioxidants should be used in HPLC. The presence of peroxides in tetrahydrofuran will have very significant effect on the baseline stability (greater than for most other organic solvents), which results in a strong recommendation always to use HPLC grade solvents which have not been stored for extended periods.
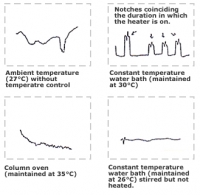
Although UV detectors are most commonly used in HPLC, refractive detectors are used when analysing compounds which do not possess UV absorption properties (such as sugars). When conducting sugar analysis using a refractive detector, baseline instability will generally become a problem. The most common reason for baseline drift problems is due to the changes in temperature of a column. Comparison of baseline stability for different temperature control methods is shown in the figures below. Baseline drift is very high at an ambient temperature without any column temperature control. Even when a water bath or a column oven is used, baseline noise can occur as a result of changes in temperature as a result of the heater switching on and off. One way to avoid this is to place the column in a stirred water bath at the ambient temperature without heating.
Comparison of baseline stability by a difference of a control method of column temperature.
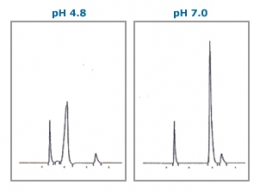
Analysis of ionic compounds by reversed-phase HPLC requires the pH of the eluent to be controlled with acid or buffering agent. The pH control stabilises the dissociation state of compounds and enhances the reproducibility of retention and separation. Unsuitable pH in analysis causes problems such as a double peaks or peak broadening. As shown in the figure below; the same compound eluted at pH4.8 shows a double peak, whilst at pH7.0 it shows a single sharp peak. When analysing an ionic compound, finding the optimum pH range for each functional group will result in avoiding such problems.
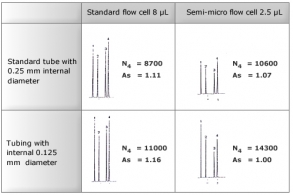
HPLC analysis of biological samples requires complicated sample preparation, depending on the component of interest. This sometimes results in very small amounts of sample being available for injection. If the injection mass is extremely small on a typical analytical column with internal diameter of 4.6mm, the component of interest might not be detected due to diffusion of substance in the column. A semi-micro column with a small internal diameter can be helpful in detecting substances in such small amounts. Such columns are also useful for saving eluent or applications involving LC/MS since lower flow rates can be selected in analytical operations.
Although the use of semi-micro columns can be useful as mentioned above, care must be taken during operations as described below. Factors causing poor column performance include dead volume of the tubing system and that of the flow cells of the detector.
The figures below show how internal diameters of tubes and internal volumes of flow cells can affect chromatograms. Starting with a tubing system compatible with analytical columns of standard sizes, column performance (theoretical plate or peak symmetry) is reduced as a result of sample diffusion outside the column. Using semi-micro columns requires suppression of this sample diffusion by the tubing system. However, it should also be noted that a small flow cell volume means a shorter path length, which in turn results in a lower peak height. It is important to optimise the system for both tubing diameter and flow cell volume, keeping in mind the considerations discussed above, in order to obtain the most appropriate column performance and detection sensitivity for the separation.
Should you be overly concerned about the degree of absorption when you set the detection wavelength as the optimum for UV absorption of the component of interest?
The absorption coefficients of some substances changes greatly with a 2 to 3nm variation in the detection wavelength. In addition, the instrumental errors can indicate a wavelength different to that actually being used for detection. In other words, however precisely you set the wavelength there may be small deviations. To avoid making such deviations having a significant effect on the results observed, it is important to choose a stable wavelength which gives little or no deviation of the absorption coefficient near the chosen wavelength rather than choose wavelength with high absorption which gives unstable results.
YMC-Pack Polymine II is highly effective in separation of a wide range of carbohydrates, including oligosaccharides and glycopeptides. It is used in normal phase separations with non-aqueous eluents or for the separation of ionic compounds by a combination of normal phase and weak anion exchange mode.
The example of analysis of ascorbic acid shown below, anions in the eluent and amino groups on the surfaces of the support material are in a state which allows formation of ion-pairs. If the ion-pairing has not yet reached ionic equilibrium, retention times of a sample can alter. YMC recommends that to enhance ion-pair formation and thereby avoid this problem, the preconditioning operation described below should be carried out prior to starting analysis.
Column preconditioning for ascorbic acid analysis (specifically for a 250 X 4.6mm I.D. column)
1) Flush the column with water at the flow rate of 1.0 mL / min for 10 minutes.
2) Flush the column with 200 mM aqueous solution of ammonium dihydrogen phosphate at the flow rate of 1.0 mL / min for 40 minutes.
3) Flush the column with water at the flow rate of 1.0 mL / min for 30 minutes.
4) Flush the column with the eluent for analytical use at the flow rate of 1.0 mL / min for 60 minutes.
However after using the eluents described above, sugar analysis using acetonitrile/water eluents can result in anomalies in peak shapes (such as peak splitting or broadening). Should this be a problem it is recommended that separate columns are used for the different analyses.

Common problems during HPLC operations include peak shape anomalies such as peak tailing and split peaks. In order to remove these problems, the cause must be precisely determined. The majority of cases are the result of inappropriate conditions of separation; including inappropriate selection of a column or solvent, or use of an old column which has a void in the packing at the top of the packing. Here we discuss the method of determining the cause of the problem with peak shapes.
The simplest way is to test the column performance according to "shipping inspection criteria" in the column inspection report which is included with every column. If the examination reveals no peak shape anomalies, the cause will be the result of inappropriate selection of separation conditions. The separation conditions, including eluent selection, will have to be revised.
However, if the initial column test reveals any anomaly, the column may be faulty. Washing the column to remove an accumulated impurity or replacement of the column is necessary. We recommend examining column performances on a regular basis and always under the identical conditions. YMC provides analytical criteria, including concentrations for test components, in column inspection reports to allow customers test the column performance of themselves.

There was a case in which the column efficiency was reduced and a peak tailing occurred (see chromatogram A) after a conventional column (150 X 4.6 mm I.D.) had been replaced with a short column (75 X 4.6 mm I.D.) to reduce the analysis time. Although this problem was corrected by replacing the injector, the cause remained unclear. The main difference between before and after the replacement of the injector was the amount of sample dispersion before the column. The shorter the retention time, the more a peak shape becomes adversely affected by pre-column dispersion.
As a result, dispersion of the sample had a greater influence on the peak shape with the short column than it had with the conventional column, resulting in deterioration of the peak shape.
Although it is often thought that problems of peak inferior shape are due to a deficiency of column performance or a defective column, some cases are caused by hardware problems introducing too much dead volume into the system. The impact of extracolumn dispersion is often overlooked when using a short column. Even with the short column, the tubing length and internal diameter, the injector type, etc should lead to minimal extracolumn dispersion.

In reversed phase HPLC, column deterioration causes poor peak shapes or reduced retention times. The column deterioration results from chemical alteration of the packing material, such as loss of, for example, C18 bonded phase or dissolution of silica-gel as the base material. Such changes result in columns which are difficult to restore and reuse.
However, use of 100% aqueous mobile phase in an ODS column can sometimes result in steep reduction in retention times for compounds as in the figure below. Many may think the reduction of this retention time is due to the permanent column damage. But this is not always the case. Sometimes the cause is believed to be the decrease in the apparent hydrophobicity of packing material due to the polarity difference between the water in the mobile phase and the surface of the packing material bonded with C18 functional groups making it difficult to solvate the phase. Overcoming this and restoring the initial retention time is easily achieved by flushing the column with 10 times its volume of mobile phase containing 50% organic solvent. This effect is believed to be the result of the decrease in the repulsion between the eluent and the C18 functional groups. If the retention time reduction occurs when using 100% aqueous mobile phase, always try to flush the column with an organic solvent/ water mixture as described above to attempt to regenerate the column.

Analysis of ionic compounds by reversed-phase HPLC is usually performed with the pH of eluent controlled using an acid or buffering agent. However, a separation using a pH range which is not optimum for the compound of interest can cause problems such as split peaks or peak broadening. Even if the peak shape is satisfactory, retention times can in some cases be non-reproducible.
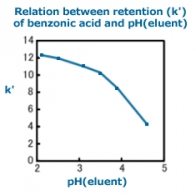
The relation between retention of benzoic acid and pH is shown in the figure below. Although the k' falls within relatively narrow limits in the pH range from 2 to 3.5, it varies widely in the pH range from 3.5 to 4.5. The pKa of benzonic acid is 4.2 and it is noticeable that the region where the k' varies most widely is near the pKa. If the eluent pH is adjusted to the region near the pKa, the result will not be reproducible because a slight change in the pH will result in a large change in the k' and have a significant effect on the separation. In fact, an eluent pH variation of just 0.1 significantly affects separation. Therefore, it is desirable that the eluent pH should be more than 1 unit away from the pKa. If the pKa is unknown, the eluent pH should be adjusted to within the region where the impact on separation seems minimal, after having carefully considered the relation between the eluent pH and the retention time.
When considering the pH value, it is also important to confirm the influence on the separation using several different eluents with their pH values slightly different from each other.
There are often reports that during the blank run as a part of a preliminary study of gradient elution without a sample, a number of peaks are detected as in figure (A). When conducting the same test but with the column was disconnected, the ghost peak disappeared as shown in figure (B). Consequently, the cause was thought to be the column.
However, despite flushing or replacing the column, the baseline would not improve. Therefore many factors other than the column were examined; the cause was found to be, in this case, the water used to prepare eluents. Standard distilled water (which is not considered suitable for HPLC) had been mistakenly used, rather than HPLC grade distilled water which leads to an excellent baseline as in figure (B).
Water purity can have a great impact on gradient elution. Even HPLC grade distilled water will become contaminated with time, causing ghost peaks. A situation which has no significant effect with isocratic elution can cause a problem in gradient elution. In gradient elution, a column is equilibrated with an eluent with low content of organic solvent so that impurities in the eluent are adsorbed and concentrated in the column. After initiating analysis, content of organic solvent increases and impurities begin to be eluted resulting in the ghost peak. The heights of ghost peaks are dependent on the duration of equilibration. The ghost peak did not appear when no column is used because of the absence of adsorption and concentration during equilibration.
In gradient analysis, solvent grade requires careful consideration especially in terms of its grade and previous storage conditions.

Although dissolving sample compounds in the eluent is the basic procedure of HPLC, with some samples this is not possible due to their solubility or the sample preparation method required. When excessive differences in solvent strength or pH between the eluent and the sample solvent arise, this can cause problems such as split peaks and broad peaks. This is considered to be caused by dispersion phenomena or varying degrees of dissociation of the analyte, as a result of the sample solvent being temporarily replaced by the eluent in the column. Examples of samples which gave this type of problem are discussed below.
The chromatogram (A) represents 2µL of an aqueous solution of a sample injected, and a shoulder is observed.
The chromatogram (A) represents 2µL of an aqueous solution of a sample injected, and a shoulder is observed.As the pH was different between the sample solution and the eluent, 4µL of this sample was injected after diluting it by a factor of 2 with the eluent to reduce the pH difference. The peak shape was much improved as in chromatogram (B).
Other ways to overcome this type of problem include reduction of the sample volume and dilution of the concentrated solution with the low strength eluent. If peak deterioration is observed, you should always confirm the sample solution has the same characteristics as the eluent. If not, you should always consider the alternatives discussed above.
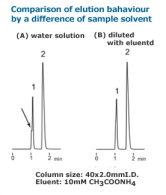
Although a column is often thought to be the cause of HPLC analyses failing to show the expected data trace, many cases can be attributed to other causes, including improper maintenance operations. This article discusses the case in which the grade of a solvent has impact on peak shapes. Here is a chromatogram of the analysis of a basic compound using acetonitrile/ water. Peak 2 represents the basic compound.
The figures on the left show the chromatograms obtained for two separations conducted under identical conditions except that the acetonitrile used was of different grades. One was HPLC grade (Figure 1); the other was reagent grade (Figure 2). Whilst the peak shape was broader with the HPLC grade acetonitrile, it was much sharper using the reagent grade. The peak shape differences can also be observed for acetonitrile from different suppliers, although they were of the same special grade. This may be because traces of impurities in the acetonitrile behave in the same way as modifiers added to an eluent. Replacing eluent with acetonitrile/ 5mM ammonium acetate produced a chromatogram similar to that in Figure 2 irrespective of the grade or supplier of the acetonitrile.
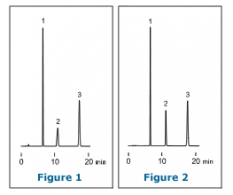
To avoid the influence of different grades, solvent specifically manufactured for HPLC should always be used. Compounds which have no dissociation groups can be analyzed with eluent containing no acid or salt, although eluents with additives such as salt should always be used when reproducibility is important.
Pressure increase is a common problem in HPLC. Possible methods to reduce pressure increase in reversed phase separation are discussed below.
If the system pressure increases, you should disconnect the column and run the system without a column to determine the line pressure. If the line pressure is high, hardware such as injectors or connection tubing may be clogged. If there is no excessive line pressure, then the high pressure may be due to the column. The column needs washing. Washing by reversed direction flow is very effective. Although generally the relative proportion of the organic solvent in the mobile phase is increased when washing, the key consideration is to choose, in accordance with the characteristics of the sample, an appropriate solvent that will dissolve easily the adsorbed material. Reversed phase separation often causes protein to be adsorbed by the packing material, resulting in high pressure. This problem can be overcome by gradient washing in the opposite flow direction with acetonitrile/ water containing 0.1% TFA, rather than washing with an organic solvent. If the high back pressre is thought to be the result of insoluble material in samples or precipitation of a sample during the separation, washing or replacing a frit (filter) might be successful.
In reality it is difficult to restore a column once it has suffered back pressure increase. It is more appropriate to prevent column pressure increases by performing sample preparation (such as protein removal), sample filtration and using a guard column.